A Comprehensive Mass Spectrometry-Based Proteomic Dataset of Human Spermatozoa
Bioinformatics Bioinformatics MethodologyPosted by mhb on 2025-12-10 14:49:08 |
Share: Facebook | Twitter | Whatsapp | Linkedin Visits: 177
Introduction
Male infertility, affecting ~7% of men globally, is rooted in germ cell defects and molecular dysregulation, yet its underlying mechanisms remain poorly understood. While assisted reproductive technologies (ART) have advanced, success rates are still suboptimal. The emergence of Data-Independent Acquisition (DIA) mass spectrometry, particularly when paired with high-resolution instruments like the Orbitrap Astral, offers unprecedented power for discovery proteomics. This study leverages this technology to construct the most comprehensive proteomic profile of human spermatozoa to date, providing a vital resource for uncovering biomarkers, therapeutic targets, and the molecular pathways underlying male reproductive disorders.
Methods
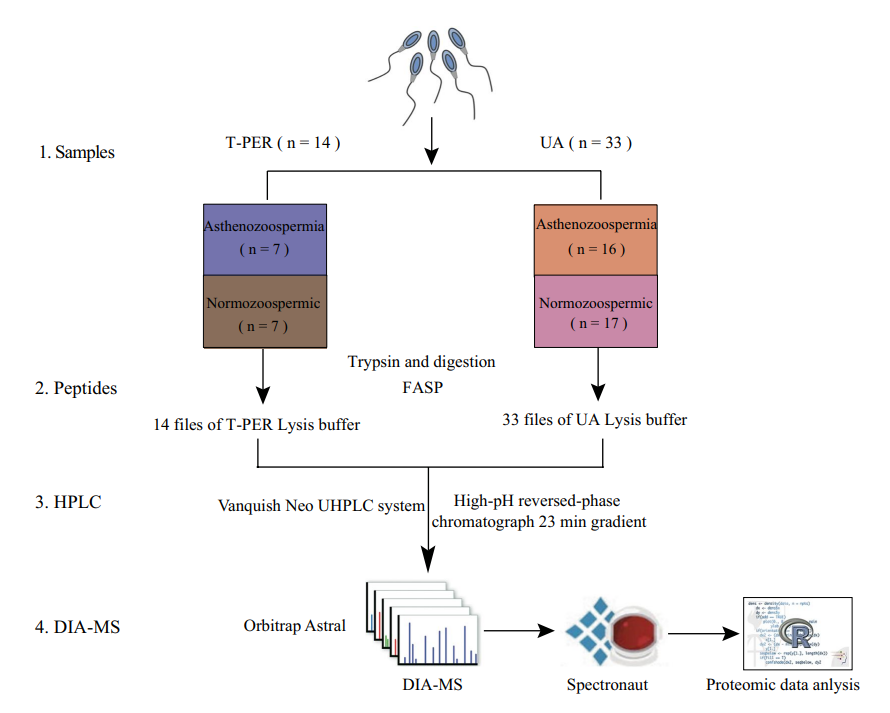
The study analyzed 47 human semen samples: 24 from normozoospermic donors and 23 from patients diagnosed with asthenozoospermia (reduced sperm motility). Proteins were extracted using two methods (T-PER and urea-assisted lysis) for comparative evaluation. Following tryptic digestion and peptide fractionation, samples were analyzed via liquid chromatography-tandem mass spectrometry (LC-MS/MS) in DIA mode on an Orbitrap Astral mass spectrometer. Raw data were processed with Spectronaut™ software (v18.7), and subsequent bioinformatics analyses included differential expression analysis, Gene Ontology (GO), KEGG pathway enrichment, and Gene Set Enrichment Analysis (GSEA).
Key Findings
Unprecedented Proteomic Coverage
Identified 9,309 protein groups, 145,355 unique peptides, and 154,062 modified peptides.
This dataset uniquely identifies 3,981 additional protein groups compared to recent major studies, representing a significant expansion in known sperm proteome coverage.
The UA (urea) lysis buffer outperformed T-PER, extracting approximately 30% more proteins with higher sequence coverage.
Proteomic Signatures of Asthenozoospermia
1,493 differentially expressed proteins (DEPs) were identified in asthenozoospermic versus normozoospermic samples.
Pathway analysis revealed that DEPs in asthenozoospermia are enriched in processes related to germ cell biology, such as intraflagellar transport, RNA splicing, microtubule-based transport, DNA replication, and cell cycle regulation.
GSEA indicated that while core spermatogenesis signatures are retained, there is significant dysregulation in inflammatory response pathways and IL2-STAT5 signaling.
Identification of Potential Biomarkers
Several proteins showed consistent and significant expression changes, including downregulation of ATRN and TNNC1 and upregulation of PIAS2.
These candidate biomarkers are associated with immune regulation, angiogenesis, and spermatogenesis, highlighting key altered physiological processes in asthenozoospermia.
Conclusion
This study establishes the most extensive and quantitative proteomic expression profile of human spermatozoa currently available, reliably detecting nearly half of all reviewed human proteins. By applying cutting-edge Astral-DIA technology, it provides deep molecular insights into the pathological state of asthenozoospermia, revealing a proteomic landscape marked by incomplete germ cell maturation and immune dysregulation. The published dataset serves as an invaluable foundational resource for the scientific community, paving the way for improved diagnostic biomarker discovery, targeted therapeutic development, and a deeper understanding of male infertility mechanisms.
Data Availability
All mass spectrometry raw files, search results, and processed data have been deposited in public proteomics repositories:
ProteomeXchange Identifiers:
PXD061698 (via iProX)
PXD061725 (via iProX)
PXD066600 (Spectronaut analysis results via PRIDE)
Publication Information
Title: A Comprehensive Mass Spectrometry-Based Proteomic Dataset of Human Spermatozoa
Authors: Ran Kong, Kaixian Yan, Defeng Liu & Wenying Wu
Journal: Scientific Data
Publication Date: 28 August 2025
Search
Categories
Recent News
- Clinical application research on the titanium metal metatarsal prosthesis designed through FEA and manufactured by 3D printing
- Manually weighted taxonomy classifiers improve species-specific rumen microbiome analysis compared to unweighted or average weighted taxonomy classifiers
- A deep learning based smartphone application for early detection of nasopharyngeal carcinoma using endoscopic images
- sCIN: a contrastive learning framework for single-cell multi-omics data integration
- CustOmics: A versatile deep-learning based strategy for multi-omics integration
- Tracking temporal progression of benign bone tumors through X-ray based detection and segmentation
- mCNN-GenEfflux: enhanced predicting Efflux protein and their super families by using generative proteins combined with multiple windows convolution neural networks
- Evaluation of normalized T1 signal intensity obtained using an automated segmentation model in lower leg MRI as a potential imaging biomarker in Charcot– Marie–Tooth disease type 1A
Popular Paper
- sCIN: a contrastive learning framework for single-cell multi-omics data integration
- CustOmics: A versatile deep-learning based strategy for multi-omics integration
- A deep learning based smartphone application for early detection of nasopharyngeal carcinoma using endoscopic images
- Tracking temporal progression of benign bone tumors through X-ray based detection and segmentation
- Clinical application research on the titanium metal metatarsal prosthesis designed through FEA and manufactured by 3D printing